A Genomic Intelligence Framework
A Genomic Intelligence Framework
Public result
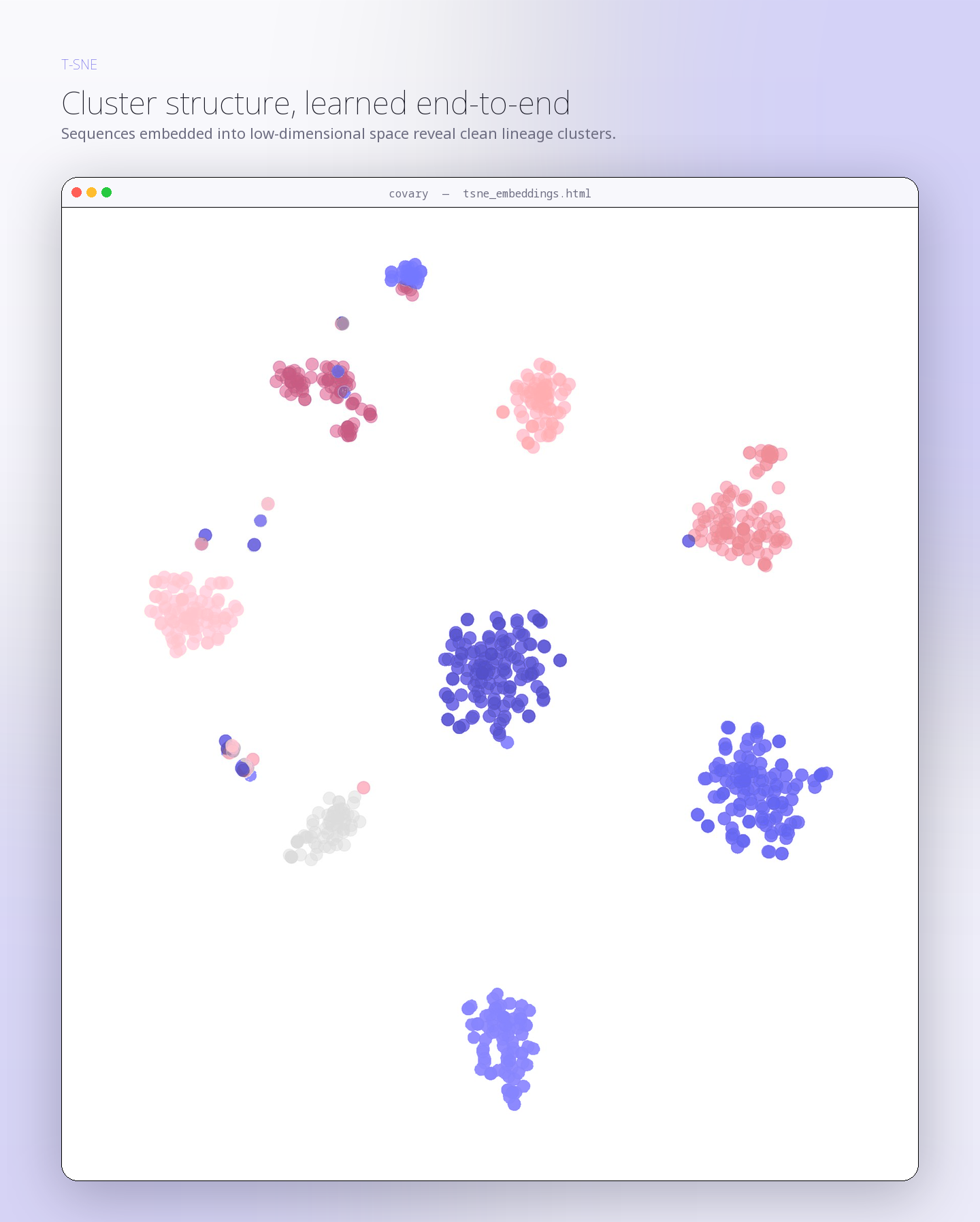
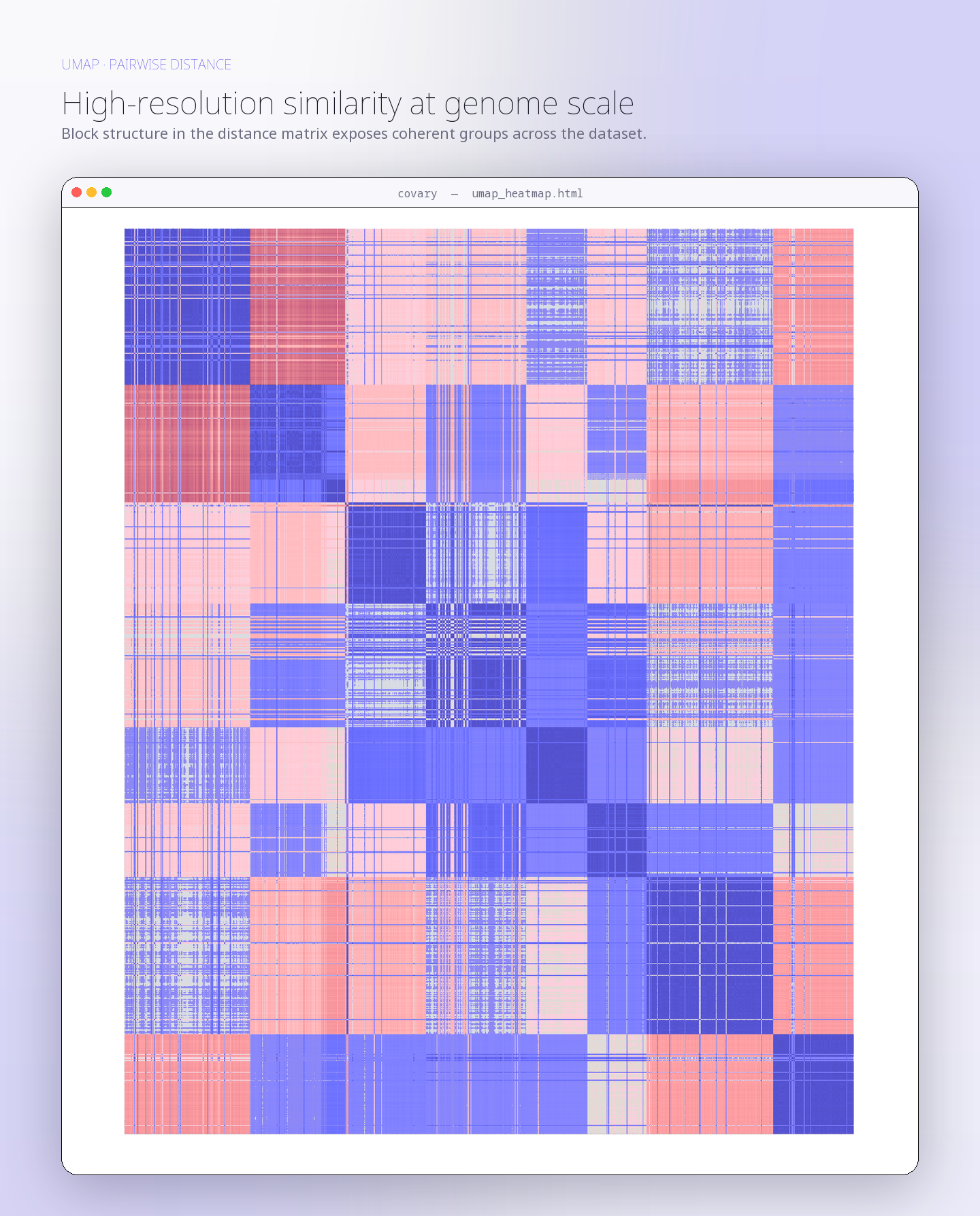
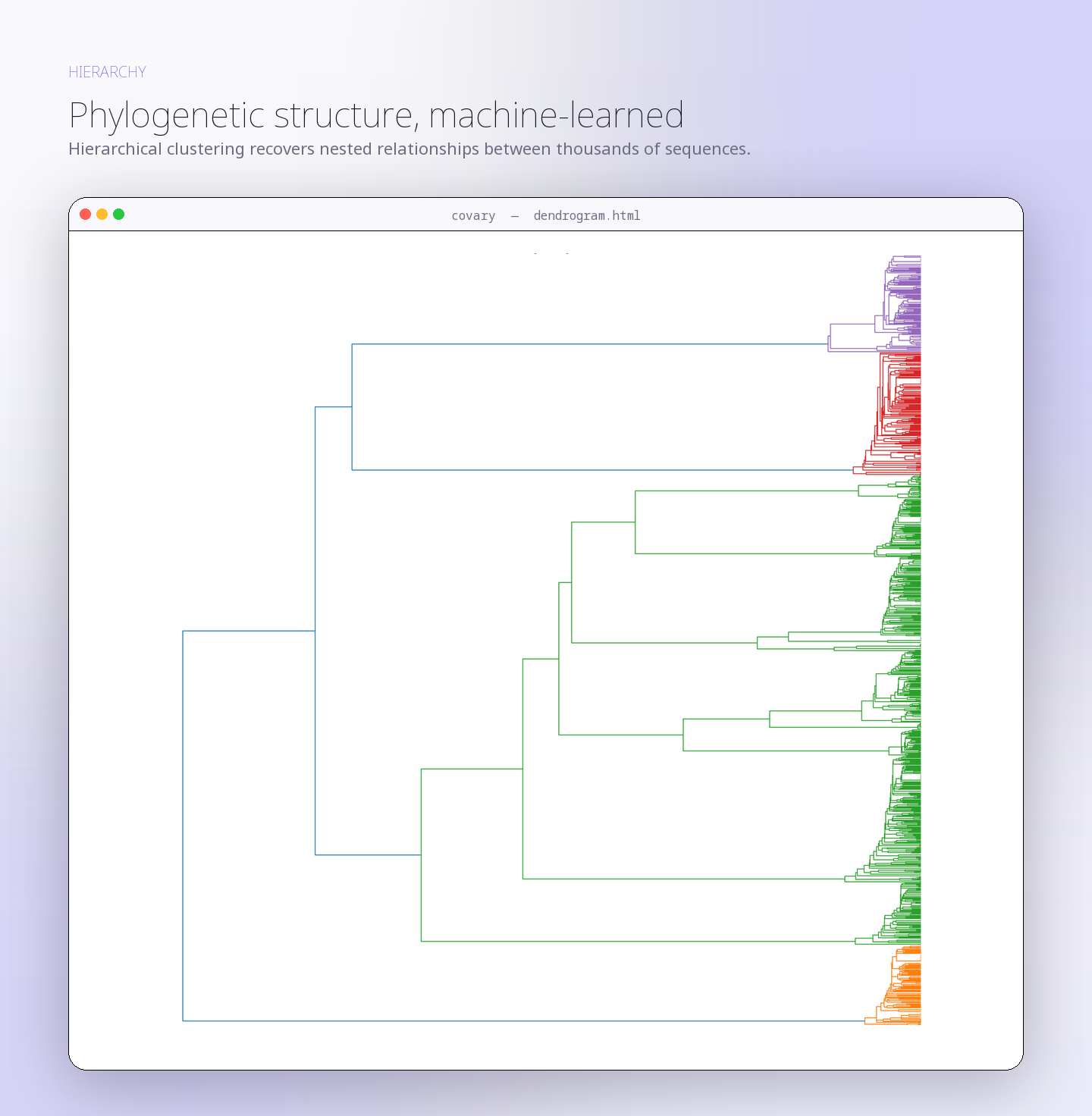
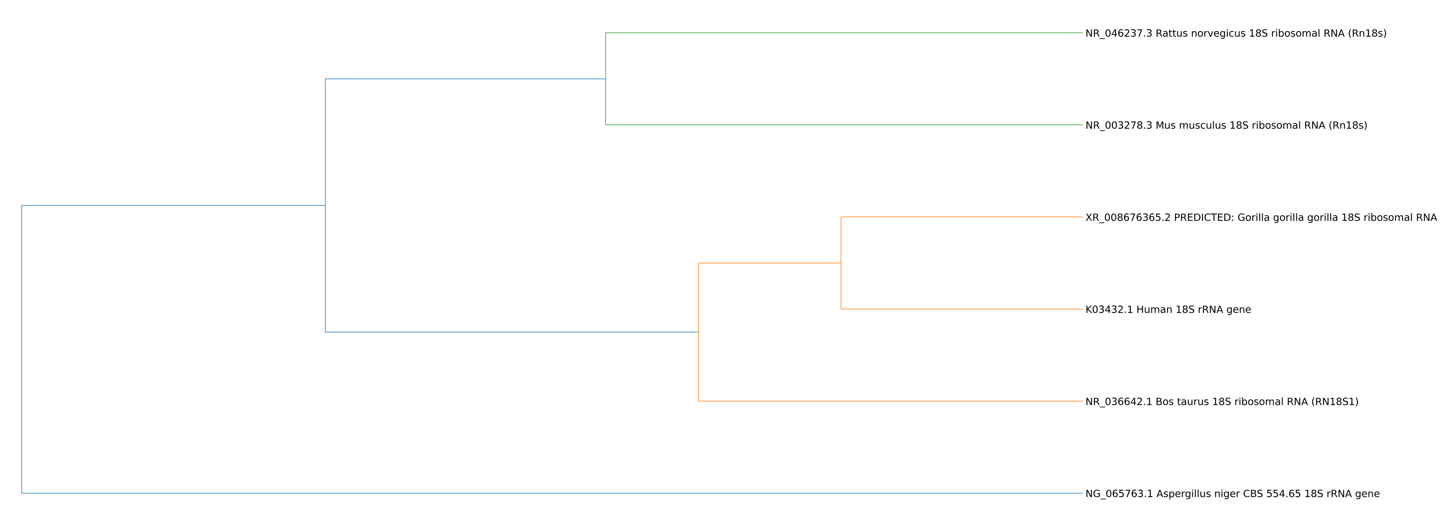
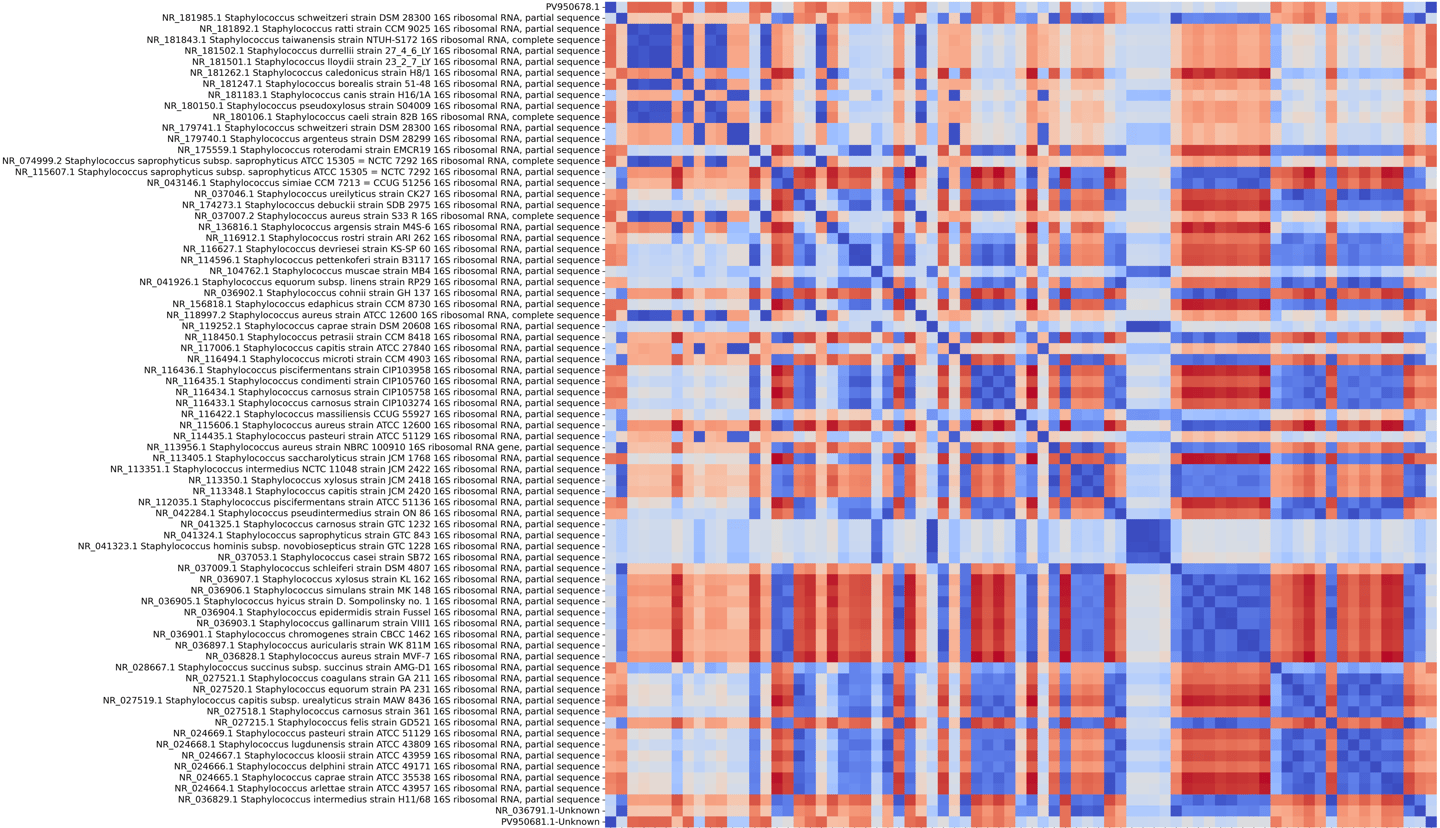
Human mitochondrial gene coalescence with bacterial homologs associated with mitochondrial ancestry
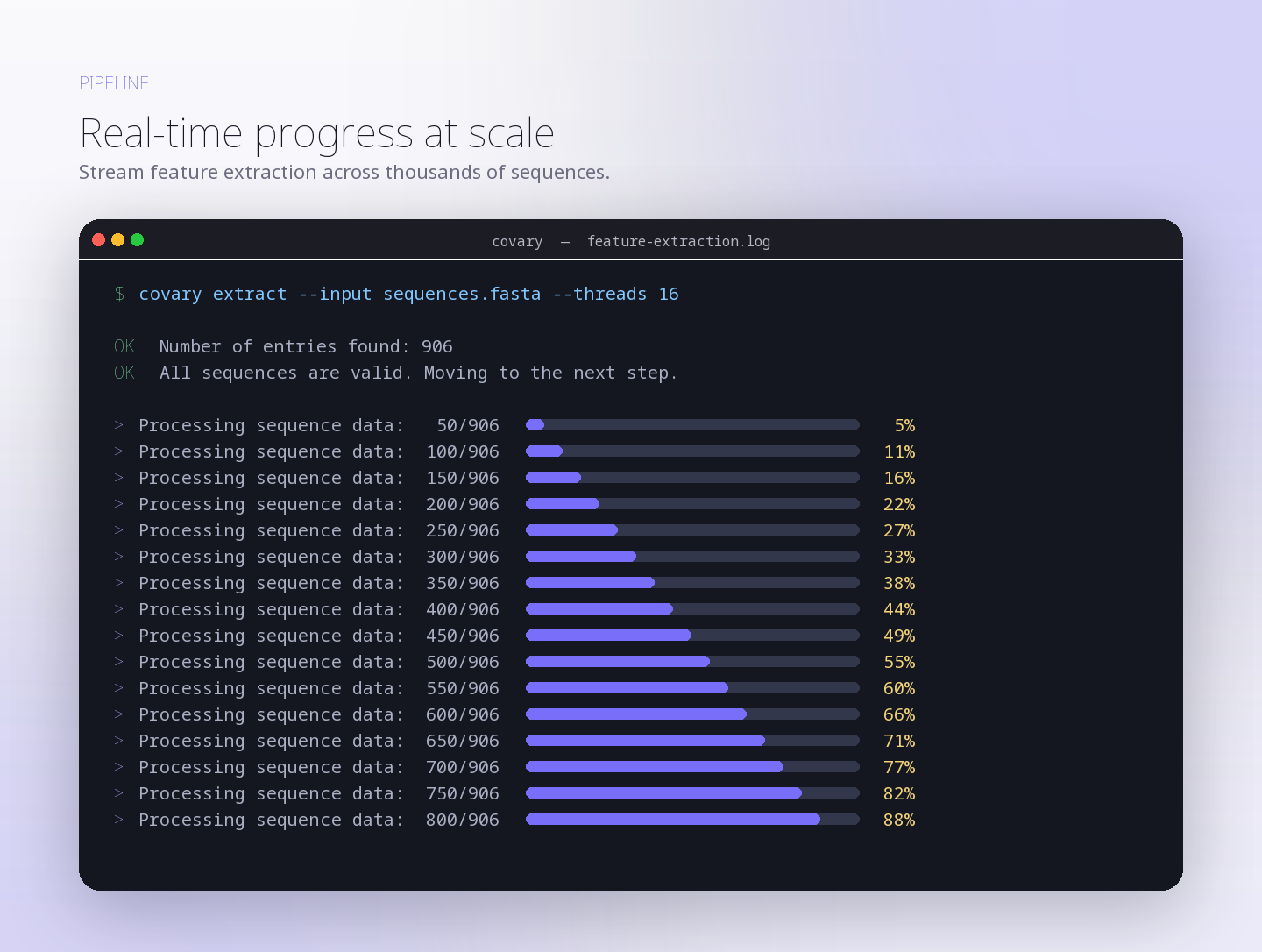
This run evaluates whether human mitochondrial protein-coding gene signatures reconstruct closer coalescence with bacterial representatives associated with mitochondrial …
User #11
ChordexBio
Open shared result →
ChordexBio